歐盟MDR與美國FDA醫療器械分類方法詳解
?
新聞資訊 ????|????
?2020-08-19 08:35:35
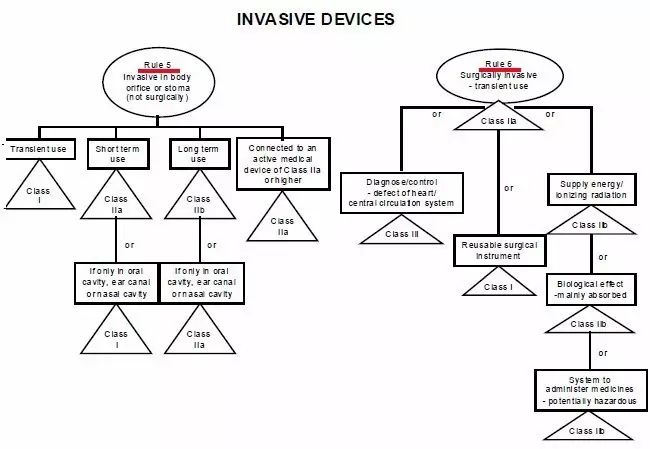
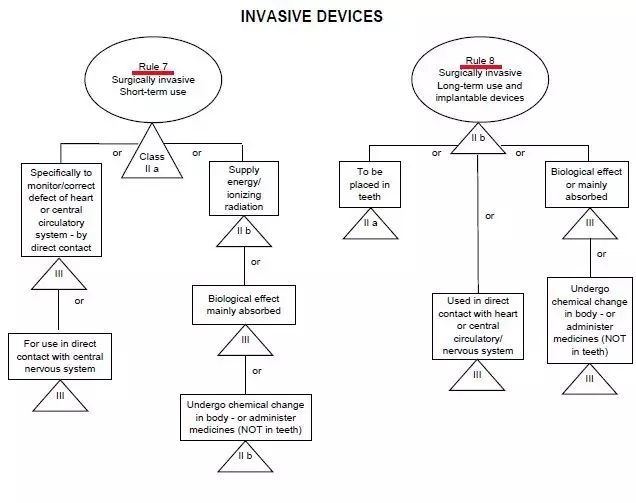
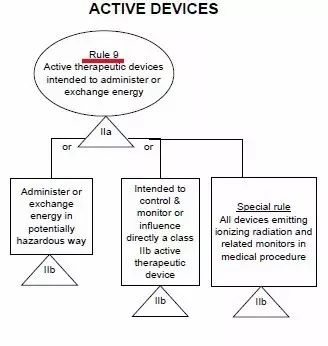
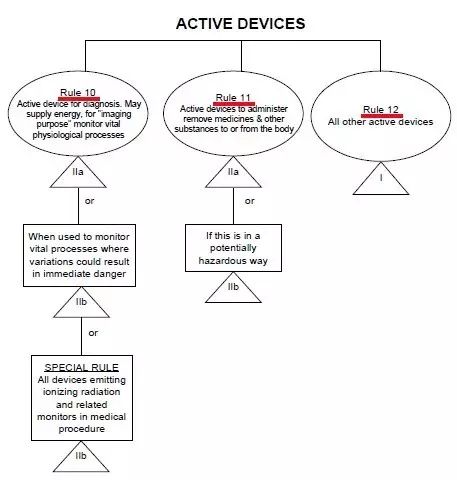
CE標志,又稱CE標記,英文為CE Marking,是一個30個歐洲國家強制性地要求產品必須攜帶的安全標志。字母"CE"是法文句子“Conformité Européene”的縮寫。其意為“符合歐洲(標準)”。一個產品帶有CE 標志(標記) 也就意味著其制造商宣告:該產品符合歐洲的健康、安全、與環境保護之相關法律中所規定 的基本要求。因而該產品是對:使用者(譯者注:人)、寵物(譯者注:家畜家禽)、財產(譯者注:物業)、及環境(譯者注:自然環境)都安全的產品。一個產品帶有CE 標志(標記) 也就是向歐盟盟國的官員表明:該產品可以合法地進入歐盟統一市場。1.一個產品帶有CE 標志(標記) 可以確保:該產品可以自由地在歐盟統一市場內流通。2.產品(必須)攜帶CE 標志(標記) (之法規)允許歐盟的: 海關依法將缺乏CE 標志(標記) 的產品在進入歐盟海關時扣留,市場監督機關依法將缺乏CE 標志(標記) 的產品從市場上取締,執法機關依法追究將缺乏CE 標志(標記) 的產品投放市場之個人或公司的法律責任。隨著越來越多的產品指令進入實施階段,越來越多的產品已經被化入必須攜帶CE標志(標記),否則不準進入歐盟統一市場流通之列。CE認證過程中判斷一個醫療器械正確的分類,僅憑器械的名稱是不夠的,必須知道完整的預期使用目的!我們經常聽到這樣的一句話問題:某某產品在CE分類里屬于幾類醫療器械?提問者也許不知道僅從一個醫療器械的名稱而判斷其CE認證過程中的分類經常是不妥當的!美國的FDA將醫療器械根據其通用的特點事先已經分類并建立了一個公開的數據庫可查詢;歐盟則是建立了一套分類規則,讓制造商根據產品的預期使用目的(Intended Purpose)按照分類規則自己進行分類。2. 同一個產品,既可以是醫療器械,也可以不是醫療器械在美國,一個產品是否為醫療器械 完全由FDA決定;在歐盟,一個產品是否為醫療器械 由制造商(申明的產品預期使用目的)決定, 比如:電熱褥既可以是醫療器械,也可以不是醫療器械。比如:制造商申明的預期使用目的不同,電熱褥既可以是I類醫療器械,也可以是II a或II b類醫療器械。4. 同一個產品,作為系統的一部分時與作為配件時屬于不同的類別比如:手術過程中用非主動式抽取腹水裝置的留在體外的盛腹水的容器,作為系統的一部分時可屬于II a類,但是作為配件時則可屬于I類。比如:X光拍片時常用的圖像儲存通信系統Picture Archiving and Communication Systems (PACS),不同制造商申明的預期使用目(功能)的不同,PACS可以是I類醫療器械,也可以是II a或II b類醫療器械。6. 類似的產品,有的屬于醫療器械MD, 有的則屬于體外診斷器械IVD 比如:采血管如果是侵入式的或接觸到皮膚的,則屬于MDD93/42/EEC 指令管轄的(普通)醫療器械MD;如果是非侵入式的或完全接觸不到皮膚的,則屬于IVD 98/79/ec指令管轄的 體外診斷器械IVD。醫療器械指令 MDD 93/42/eec附錄九中詳定18條規則,按醫療產品的危險程度,將產品分為Ⅰ類、Ⅱ a類、Ⅱ b類、Ⅲ類。2、如果器械是和其它器械配合使用,分類規則分別適用于每種器械;規則1~4、所有非創傷性器械均屬于I類,除非他們:于II a類或更高類型的有源醫療器械類 II a類與中央循環系統(CCS)或中樞神經系統接觸的器械 III類診斷/監視體內放射藥物分布 II a類 (r照相機、正電子發射成像儀)危險情況下監視生理功能 II b類 (手術中的血氣分析儀)規則11、控制藥物或其他物質進出人體的有源器械II a類(吸引設備、供給泵)如以一種潛在危險方式工作II b類 (麻醉機、呼吸機、透析機、高壓氧艙)(觀察燈、牙科椅、輪椅、牙科用治療燈、記錄處理觀察診斷圖象用的有源器械) 規則13、與醫用物質結合的器械(含殺精子的避孕套、含抗生素的牙髓材料) III類規則14、避孕用具(避孕套、子宮帽 II b類 ) II b/III類 (子宮內避孕器 III類)規則16、用于記錄X射線圖象的器械(X光片) II a類規則17、利用動物組織的器械(生物)心臟瓣膜、腸線、膠原) III類
FDA醫療器械分類方法詳解
美國FDA醫療器械產品目錄中共有1700多種。根據風險等級的不同,FDA將醫療器械分為三類(Ⅰ,Ⅱ,Ⅲ)。風險等級逐級升高,Ⅲ類風險等級最高。FDA對每一種醫療器械都明確規定了其產品分類和管理要求。首先,對于任何產品,企業都需進行企業注冊與產品列名(Enterprise Registration and Product Listing)。絕大部分產品只需進行注冊、列名和實施GMP規范,產品即可進入美國市場(其中極少數產品連GMP也豁免,極少數保留產品則需向FDA遞交510(K)申請即PMN(Premarket Notification))這些器材只要經過一般控制就可以確保其功效與安全性,如拐杖、眼鏡片、膠布等,約占全部醫療器材的27%。實施GMP(Good Manufacturing Practice)。一般控制 + 特殊控制(Special Control)企業在進行注冊和列名后,這些產品除了上述一般控制之外,其余大多數產品均要求進行上市前通告(PMN:Premarket Notification)(即510K)。少量的II類產品可以豁免上市前通告程序。生產企業須在產品上市前90天向FDA提出申請,通過510K審查后,產品才能夠上市銷售。一般控制 + 上市前許可(Premarket Approval)企業在進行注冊和列名后,須實施GMP并向FDA遞交PMA申請(部分Ⅲ類產品還是PMN)。一般來說, III類產品多為維持、支持生命或植入體內的器材,對病患具有潛在危險,可能引起傷害或疾病者,如心律調節器、子宮內器材及嬰兒保溫箱等,約占所有器材的8%。這些器材必須取得FDA的PMA之后方能銷售。對Ⅰ類產品,企業向FDA遞交相關資料后,FDA只進行公告,并無相關證件發給企業;
對Ⅱ、Ⅲ類器械,企業須遞交PMN或PMA,FDA在公告的同時,會給企業以正式的市場準入批準函件(Clearance),即允許企業以自己的名義在美國醫療器械市場上直接銷售其產品。
至于申請過程中是否到企業進行現場GMP考核,則由FDA根據產品風險等級、管理要求和市場反饋等綜合因素決定。
綜合以上內容可知,絕大部分產品在進行企業注冊、產品列名和實施GMP,或再遞交510(K)申請后,即可獲得FDA批準上市。除了依據器材的風險來分級之外,依照器材的用途,FDA把現有醫療器材產品總共被分成16類(medical specialty "panels")。 | Regulation Citation (21CFR) |
| | |
| | |
| | |
| | |
| | |
| Gastroenterology and Urology | |
| General and Plastic Surgery | |
| | |
| | |
| | |
| | |
| | |
| Obstetrical and Gynecological | |
| | |
| | |
| | |
| | |
| | |
| | |
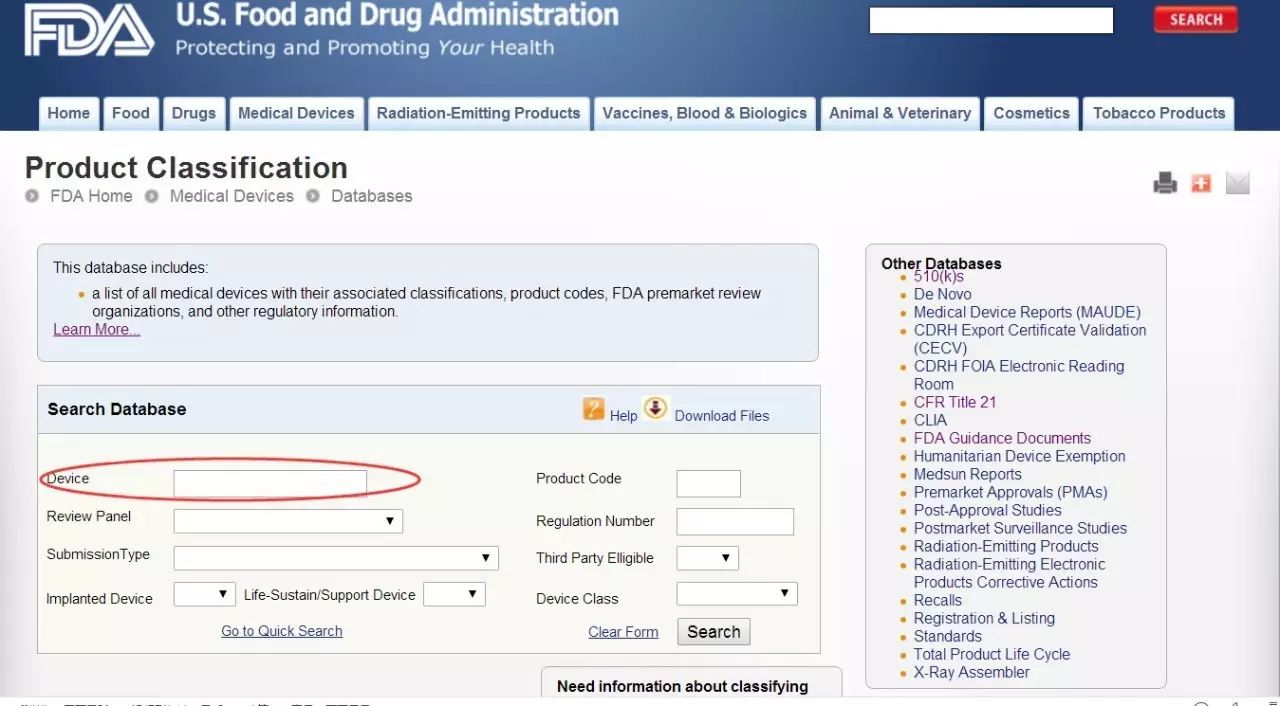
當然,這只是一個大致的分類。具體到某一個器械,屬于哪一類,以及是否得到豁免,均需通過查找其相對應的法規編號。1700類產品,每一類產品都有一個法規編號與之對應。方法一:進入FDA官網中的classification database頁面(http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpcd/classification.cfm)。您就會看到下方的頁面:有兩種方法。
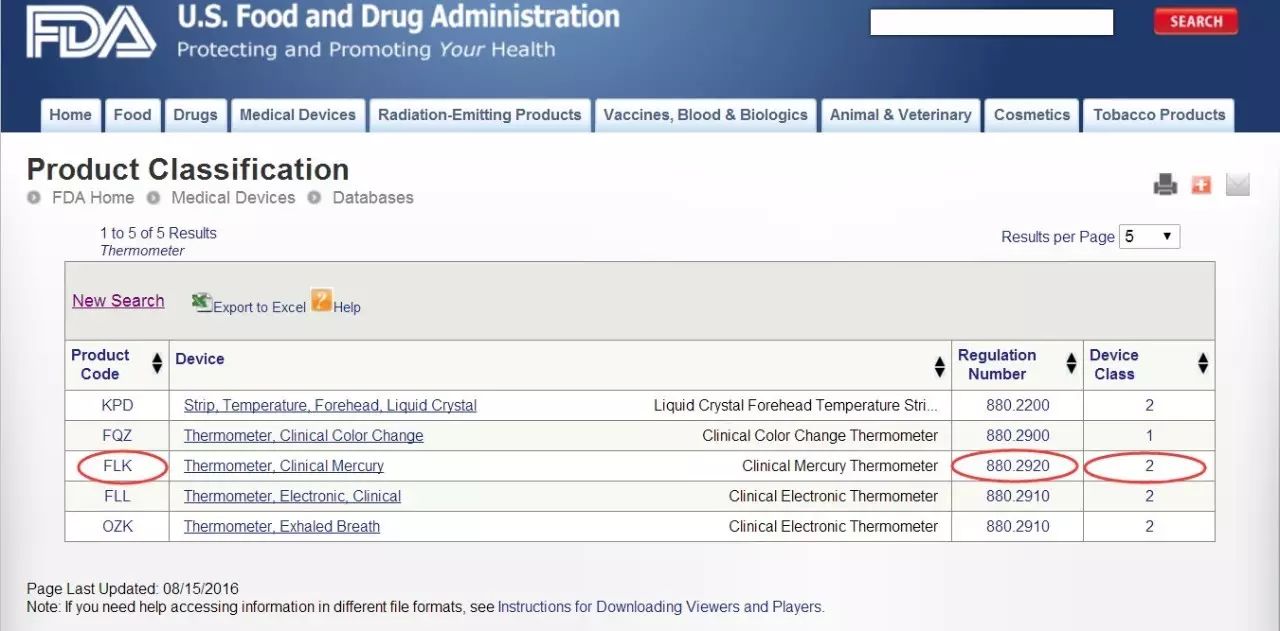
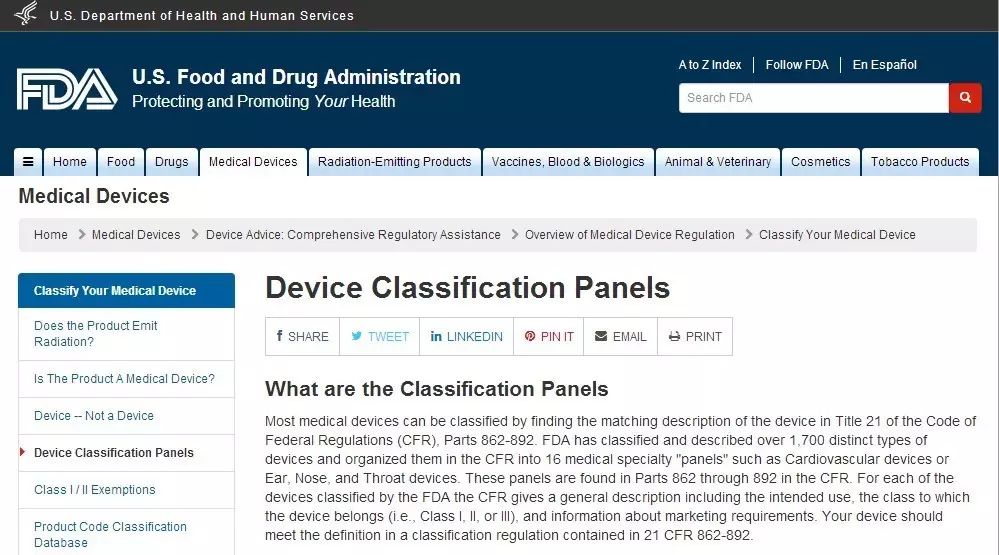
如果您知道器械的部分名稱,您就可以找到相對應的法規編號。比如:要找 Clinical Mercury Thermometer這個產品的法規編號,就把Thermometer輸進紅框中,接著就可以看到下面這個頁面。在這里,不僅能夠看到法規編號,還可以看到產品代碼和器械分類級別。方法二:如果知曉該器械屬于之前所提及的16類中的某一類,就可以進入FDA官網的Device Classification Panels(http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/Overview/ClassifyYourDevice/ucm051530.htm)頁面搜索。
更多檢測內容 電話咨詢:400 880 4601 / 156 6560 3132(微信同號)
【來源:internet】
版本與免責申明:
①本網凡注明“來源:xxx(非本網)”的作品,均轉載自其它媒體,轉載目的在于傳遞更多信息,并不代表本網贊同其觀點和對其真實性負責,且不承擔此類作品侵權行為的直接責任及連帶責任。
②如涉及作品內容、版權等問題,請在作品發表之日起兩周內與本網聯系,我們將在第一時間刪除內容。
本文標簽:醫療器械